

Is Wuhan a Red Herring? Addendum
Addressing some criticism.

Let’s not beat around the bush and get to the main point which is in the paper by Kumar et al.
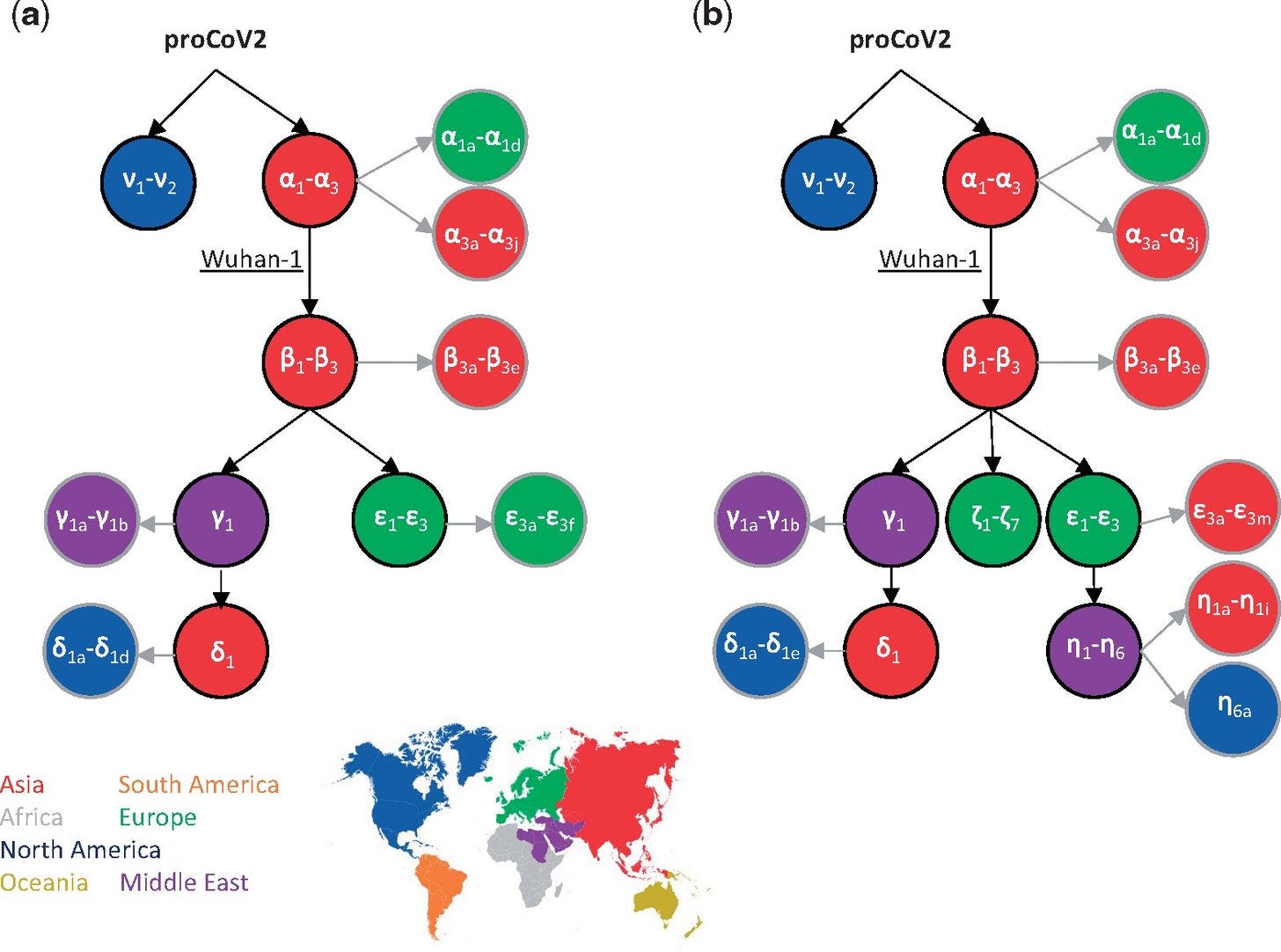
According to the mutational history, the Wuhan-1 strain evolved by three successive α mutations (two synonymous and one nonsynonymous) in proCoV2 (α1, α2, and α3). This progression is statistically supported (BCL = 100%), which is made possible by the presence of 896 intermediate genomes containing one or two α variants in the 29KG data set. Importantly, three closely related nonhuman coronavirus genomes (bats and pangolin) all have the same base at these positions as does the proCoV2 genome, suggesting that the ancestral genome did not contain α variants. Furthermore, genomes with ν variants of proCoV2 do not contain the other 47 variants, all of which occurred on the genomic background containing α1–α3. These facts support the inference that coronaviruses lacking α variants were the ancestors of Wuhan-1 and other genomes sampled in December 2019 in China (fig. 1c). Therefore, we conclude that Wuhan-1 was not the direct ancestor of all the early coronavirus infections globally.
Which pill would you take the red pill or the blue pill?

Let me rephrase this excerpt above from the paper.
Ancestral genomes DO NOT have α mutations.
The ν variant [identified in North America] is closer to the ancestral SARS-CoV2 genome than the α variant [identified in China].
There has been some criticism of this Substack on the origins of SARS-CoV2 and I would like to respond to some of the questions that people have had in order to clear up some misunderstandings and maybe rephrase the main findings.
One source of contention that some have was whether EVALI is COVID-19. We don’t have proof of this for the proposed dates spanning April to September — as a novel pathogen was never identified because there was never an intention to look for one. You can’t create a PCR test nor an antibody test without having the genome for said virus.
Was there transmission associated with EVALI? We don’t have any direct evidence other than a news story from ABC:
Michael Gasaway, principal of Madison Consolidated High School (MCHS), said the teachers fell ill while monitoring an area in the school’s C-wing. They were taken to hospital as an apparently precautionary measure along with three other students who also displayed minor symptoms.
If EVALI was a wild type circulating virus it didn’t have the mutations necessary to create the kind of pandemic we are all too familiar with. Transmission within the vaping cohort could have happened mouth-to-mouth from the sharing of devices, which young people tend to do. High death rate per vaping user of older individuals was observed in EVALI cases which gives us an indication that there could have been an age stratified risk associated with the lung injury.
One interesting graph that had me puzzled is this one.

If I recall, when making a batch of vaping liquid you normally make quite a lot to — save time and money — while ensuring proper ratios are adhered to of the various ingredients that compose eliquid or illegal THC cartridges. Now if we take what the current narrative tells us, which is that EVALI was caused by vitamin e acetate or some other chemical in brand name or illegal cartridges/e liquids, then why does this graph strongly resemble a pathogen outbreak with an exponential curve and then a slow taper. Wouldn’t a huge number of people experience adverse events as soon as they started vaping?
The only difference that we seem to observe between EVALI and COVID-19 is when we look at the case definition of EVALI.

Difference
Using an e-cigarette(“vaping”) or dabbing in 90 days prior to symptom onset.
Negative respiratory viral panel and influenza PCR or rapid test.
Wow! Definitely can’t see any issues here with SARS-CoV2 false negatives after it’s on the scene.
2019-2020 flu season was pretty bad and early!



But when is week 40?
Beginning of October…
Please comment if you felt like you had Covid in the summer of 2019 or know someone who has.
Daoyu has been saying that Csabai's Antarctic reads contain C29095T, but actually there's just 3 reads with C29095T, and the bases with the mutation all have a very low quality: quality 6 in base 137 of read 5599415 in SRR13441704_2 (where there's 14 remaining bases after it in the read out of which half are different from the reference), quality 5 in base 94 of read 21585669 in SRR13441708_2 (where 2 bases before it there's another base that is different from the reference that has a quality of 10), and quality 5 in base 2 of read 23910570 in SRR13441708_2 (where the first base of the read is also different from the reference). And a Phred33 quality of 5 corresponds to an expected error rate of 1/10^.5, or about 32%. So I think the 3 instances of C29095T in the Antarctic reads are just sequencing errors.
`samtools mpileup` doesn't include bases with quality below 13 by default, so in order to see the 3 mutations, you have to remove the quality cutoff with `samtools mpileup -Q0`:
brew install parallel sratoolkit bowtie2 samtools
printf %s\\n SRR134417{00..11}|parallel -j12 fastq-dump --split-3 --gzip
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta&id=MN908947.3' >sars2.fa;bowtie2-build sars2.fa{,}
for x in SRR134417*.fastq.gz;do bowtie2 -p3 -x sars2.fa -U $x --no-unal|samtools sort -@2 ->${x%%.*}.sars.bam;done
for x in {00..11}_{1,2};do samtools mpileup -f sars2.fa -Q0 SRR134417$x.sars.bam|ruby -alne'next unless[29095].include?($F[1].to_i);next if$F[3]==0||$F[4]=="*";t=$F[4].upcase.gsub(/[.,]/,$F[2]).gsub(/[+-]\d+[A-Z]+/,"").chars.tally;puts [$F[1],$F[2],t["A"]||0,t["C"]||0,t["G"]||0,t["T"]||0]*" "'|sed s/^/$x\ /;done|sort -nk2|(echo run pos ref A C G T;cat)|column -t
The sample Guangdong/FS-S30-P0052/2020 is also interesting, because it has only 5 mutations from Wuhan-Hu-1 but all of them are shared with RaTG13. The sample has all 3 proCoV2 mutations and the 2 additional mutations C21711T and C24382T. However the Antarctic reads don't seem to include either of the additional mutations. In a set of 719,679 GISAID sequences with collection date in 2020 or earlier, if sequences with over 100 mutations are excluded which are mostly other SARS2-like viruses, then C21711T is found in 128 sequences and C24382T is found in 1,156 sequences: `curl -s https://cdn.discordapp.com/attachments/1093243194231246934/1129149184939929713/early.comb.xz |gzip -dc|sort -k4>early.comb;awk -F\\t '$11<100' early.comb|cut -f12|grep -o '[A-Z]21711[A-Z]'|sort|uniq -c|sort`.
For some reason many of these interesting mutations seem to pop up at Guangdong before Hubei, even though GISAID has much more early samples from Hubei than Guangdong. On GISAID the 7 C29095T samples with the earliest collection date are also found at Guangdong: `grep C29095T early.comb|grep Human|cut -f1,3-7,11,12-|head|tr \\t \|`.
In Jesse Bloom's paper about the recovered deleted sequences, in the tree where he selected WA1/proCoV2 as the root, there's only 3 first-level branches under the root, out of which one branch consists of the mutations C865T and C13694T. Both mutations are common in the Antarctic reads, but there's only 4 samples on GISAID with a collection date in 2020 that contain both mutations: 2 samples from Singapore from late January and 2 samples from Guangdong from early February. All 4 samples have only 5 mutations from Wuhan-Hu-1, where the other 3 mutations are the mutations of proCoV2/WA1. I came up with the term "Guangdong-Singapore line" to refer to the line with the C865T and C13694T and mutations. Neither of the mutations seems to be ancestral though. But it's still interesting that the mutations are found in early Chinese samples without C18060T, so there seems to be an overlooked sublineage of proCoV2 which is also found in the Antarctic reads.
When I did variant calling for the pooled reads from all Antarctic runs, I got a high MAPQ value for the mutation A16156G, which is included in only one sample at GISAID with a collection date in March 2020 or earlier, which is a sample from Guangdong with a total of 4 mutations relative to Wuhan-Hu-1. Two of the additional mutations are the lineage A mutations, and the fourth one is T9013A, which is a rare mutation which is not found in any other samples at GISAID until a sample from Brazil with a collection date in May 2020.
C23525T is another rare mutation in the Antarctic reads that got high MAPQ for my pooled reads. It's found in 6 samples at GISAID with a collection date in February 2020 or earlier, but one of the samples is from Guangdong, and its only other mutation relative to Wuhan-Hu-1 is C13694T which is one of the two "Guangdong-Singapore" mutations.
Out of the Antarctic mutations with a fairly high MAPQ, the only mutations which appear to be ancestral are the 3 proCoV mutations, but other mutations appear to not be ancestral: T9440A, C13694T, A16156G, A17039G, A18082G, A18082G, A21975C, C23525T, G23525T, C25498T, C26895T, and G29449T. So I think it's possible that the Antarctic reads don't actually contain strains that are more ancestral than WA1.
When I ran `samtools mpileup` for the Antarctic reads with the default quality cutoff of 13, I got 20 reads which included position 3037 but zero of them had C3037T.
- Thesis of Toxic substance (VEA) present then removed:
Compare with the curve for the 1981 Spanish Toxic Oil Syndrome, page 7 of this doc:
http://www.euro.who.int/document/e84423.pdf
It does not have the slow curve up-front: a toxic batch has a massive up-front effect, which then fizzles off. The Toxic Oil was identified years later.
Yes EVALI was cover-up for early COVID.
We note who insists that it's not, with a level of scrutiny/exploration/data much different from what is applied to WIV/Wuhan-CDC or even Fauxi/Sadsack/Baryc.
What's at play here? Directed Limited Hang-out?
Who died unexpectedly, when?
When did it spread?
- 2009 "Swine" H1N1 Flu is clearly high & out of sync/very early.
Was that a rehearsal, or also a leak? a first attempt *also with a pandemic declaration*, but that fizzled too fast, and also a deadly vakscene withdrawn after maiming a couple hundreds?
The next big ILI curve is 2017-2018 cyan, better synchronized - while it was NOT in Europe
Page 4 of https://www.ecdc.europa.eu/sites/default/files/documents/AER_for_2019_influenza-seasonal.pdf
2017 first invisible contamination wave.